

2.5 Trial results: whole trial population
Page last updated: September 2016
Information Requests
- Present the results on effectiveness for each trial for relevant outcomes (Subsection 2.5.1)
- Present adverse event data (Subsection 2.5.2)
- Cross-reference the source documents (Subsection 2.5.3)
Report the results from the studies for the whole trial population in this subsection. Additional analyses – such as subgroup analyses, meta-analyses, indirect comparisons or adjustments for treatment switching – are presented in Subsection 2.6.
In some cases, the results for the whole trial population will be presented in Subsection 2.6 as part of the additional analysis. If this is the case, cross-reference to the relevant tables in Subsection 2.6 but interpret the results (in the context of the nominated MCID, where applicable) for the whole trial population for each trial in Subsection 2.5.
2.5.1 Effectiveness
For each trial identified in Subsection 2.2, present the results of the primary outcome, and other relevant outcomes identified in Subsection 2.4, for the whole trial population.
In general, present the following details (where permitted by the data):
- the number of patients at risk or providing data to the results
- the number of patients experiencing the event (if appropriate)
- the percentage of patients with the event, and means (standard deviation) or medians (interquartile range) within groups as appropriate
- CIs of the outcomes within groups
- relative and absolute differences between groups, and CIs
- an interpretation of the results
- a discussion of the results in the context of the nominated MCID
- a statement of whether the results are used in an economic evaluation in Section 3.
Tables 2.5.1–2.5.3 show examples of how to present the different types of data.
Although the outcomes are defined in Subsection 2.4, it is important to present the timing of the outcome assessment (eg EORTC-QLQ C30, change from baseline at six weeks) in the table heading or as a footnote to the table. If there are multiple studies that differ in timing of the measurement of the outcome or length of follow-up over which the outcome can be observed, present these differences below each results table. Justify and discuss any early stopping of a trial or reliance on interim analysis in the interpretation of the results.
Dichotomous data
|
Trial ID |
Proposed medicine |
Main comparator |
Relative risk (95% CI) |
Risk difference (95% CI) |
|---|---|---|---|---|
|
Trial 1 |
n/N with event (%) |
n/N with event (%) |
[add] |
[add] |
|
Trial 2 |
n/N with event (%) |
n/N with event (%) |
[add] |
[add] |
|
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
CI = confidence interval; n = number of participants with event; N = total participants in group
Continuous data
Many trials measure a continuous variable at baseline and again at a prespecified time point. The treatment effect from such trials can be reported in several ways. Analysis of covariance (ANCOVA) is the most commonly used general approach, but other approaches might also be acceptable. The usual output from ANCOVA is the difference in mean change scores, adjusted for baseline scores. Report these in Table 2.5.2. Where statistical control has been applied (eg ANCOVA), report and justify the covariates used and the assumptions required for the approach (and how they were tested), and discuss the effect of controlling for covariates on the estimated comparative treatment effect.
If the outcome was measured at more than one time point, justify why that end point was selected. Discuss whether the treatment effect differs across other time points, and present these results in an attachment, or provide a clear reference to where they are presented in the sponsor’s study report.
|
Trial ID |
Proposed medicine (mean values) |
Proposed medicine (mean values) |
Proposed medicine (mean values) |
Main comparator (mean values) |
Main comparator (mean values) |
Main comparator (mean values) |
Mean difference (95% CI) |
ANCOVA (95% CI) |
|---|---|---|---|---|---|---|---|---|
|
Trial 1a |
Baseline (SD) |
End point (SD) |
Change (SD) |
Baseline (SD) |
End point (SD) |
Change (SD) |
[add] |
[add] |
|
Trial 2a |
[add] |
[add] |
[add] |
[add] |
[add] |
[add] |
[add] |
[add] |
|
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
ANCOVA = analysis of covariance; CI = confidence interval; SD = standard deviation
a For each trial, state the number of participants in the group and the number reporting data for each time point.
Where continuous data are translated to dichotomous data in the economic evaluation or to support the clinical claim, justify the use of the threshold to convert the data. If the threshold is not well supported by the literature, present sensitivity analyses using different thresholds, or present a cumulative distribution function of the continuous outcome separated by treatment arm. Clearly show the effect of the choice of threshold to determine the dichotomous outcome on the comparative treatment effect.
Time-to-event data
|
Trial ID |
Proposed medicine |
Proposed medicine |
Main comparator |
Main comparator |
Difference in median |
P value (log rank test) |
Hazard ratio (95% CI) |
|---|---|---|---|---|---|---|---|
|
Trial 1 |
n/N with event (%) |
Median time to event (95% CI) |
n/N with event (%) |
Median time to event (95% CI) |
[add] |
[add] |
[add] |
|
Trial 2 |
[add] |
[add] |
[add] |
[add] |
[add] |
[add] |
[add] |
|
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
[etc] |
CI = confidence interval; n = number of participants reporting data; N = total participants in group
Present relevant Kaplan–Meier curves for each included study. If the sponsor cannot access the study data or cannot request a Kaplan–Meier curve, and it has not been published, clearly state this.
Describe the method for analysing the time-to-event data. State any assumptions and how they have been tested. For example, where the analysis is based on a Cox proportional hazards model, present the hazard ratios and their 95% CIs. Discuss whether the results are consistent with the assumption of constant proportional hazards. Present results of testing for proportional hazards. Where the assumption of constant proportional hazards is not reasonable, present alternative methods for estimating comparative effectiveness. Where restricted mean survival time is used, present estimates of the restricted mean (and the difference in restricted means) calculated at several time points over the duration of the trial.
Ordinal or categorical data
Attempt a similar approach as the method described for continuous data if the trial results are available as ordinal or categorical data (eg a Likert scale for patient-reported outcome measures). Expert biostatistical advice will be helpful in such circumstances, particularly to meta-analyse the data.
Multiattribute utility instrument data
Report MAUI results (with 95% CI) for each time point and each arm within the trial. Report the number of patients eligible for the questionnaire and the number of patients who responded for each time point. Where this cannot be done, explain why and present the results as specified in the trial protocol. Report the difference between the arms (with 95% CI) as the integrals between the mean utility weights obtained over time up to the median (or other relevant time point) follow-up in the trial. If an alternative approach for comparing MAUIs was used, explain how this was done.
If the scoring algorithm has not been derived from the general population in Australia, consider presenting a sensitivity analyses using alternative scoring algorithms, if possible. If more than one MAUI has been used in the included study, compare the results from the two MAUIs.
Discuss the interpretation of these results. Assess the results against other outcomes measured in the trial. In particular, discuss the consistency or inconsistency with any concomitantly assessed disease- or condition-specific patient-reported outcome measure and/or generic patient-reported outcome measure.
Effectiveness in the context of minimal clinically important difference
Discuss the results of the primary outcome and main patient-relevant outcome with reference to the MCID. Also follow this guidance for analyses presented in Subsection 2.6.
State whether the intervention group has achieved a difference as large as or larger than the proposed MCID when compared with the comparator group. Comment on the extent to which the CI for the comparison includes differences smaller than the proposed MCID.
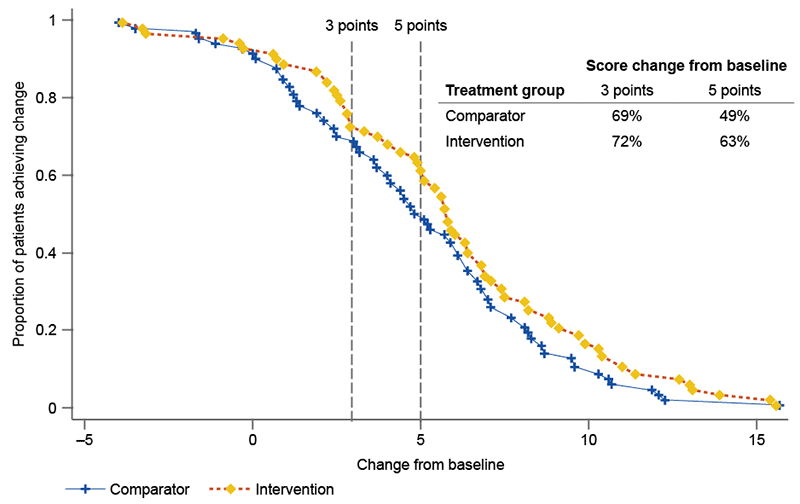
In addition to the analysis above, where continuous or ordinal outcomes (eg patient-reported outcome measures) can be presented as a responder analysis, present such an analysis. Present a cumulative distribution function (see example in Figure 2.5.1). Compare the number of patients in each arm that achieved a response greater than the proposed MCID (derived in Section 2.4.4) using a relevant statistical test and for alternative values of the MCID, where possible.
Figure 2.5.1 Cumulative distribution function
Applying a noninferiority margin
Compare the least favourable tail of a 95% CI with the noninferiority margin and determine whether the ‘worse’ result would be regarded as noninferior. Assess this using both intention-to-treat and per-protocol approaches. Discuss discrepancies between the approaches. Where one approach is not available, discuss whether the approach may have resulted in a different conclusion.
Important differences are usually presented as absolute differences, whereas trial comparisons are usually done using relative measures. Explain how the noninferiority margin is converted from one outcome measure to another, if necessary.
Discuss possible reasons if noninferiority cannot be concluded. Discuss other considerations that may support the conclusion of noninferiority (eg whether the medicines are of the same class, the point estimate favours the proposed medicine, whether there are safety or tolerability advantages of the proposed medicine).
Explain and justify any alternative approach to establish noninferiority to that described above and ensure it clearly tests that the proposed medicine is superior to placebo and is not inferior to the proposed comparator by an important extent.
2.5.2 Adverse events
At a minimum, the following categories of adverse events should be reported:
- any adverse event
- any adverse event resulting in discontinuation of the randomised treatment
- any serious adverse event18
- any adverse event resulting in death
- each and every other type of adverse event where the frequency or severity differs substantially across groups, for each study listed in Subsection 2.2.
Where additional adverse events are to be reported (eg treatment-emergent adverse events, adverse events of special interest), explain the importance of the adverse event and interpret the result.
Report adverse event data as both the number of patients reporting an adverse event in each category and the absolute number of adverse events in each category. The absolute number of events in each category may be a more appropriate estimate for costing adverse events in an economic or financial analysis, rather than the number of patients who experience an adverse event, because the latter will not capture patients who experience two events in the same category.
For each important adverse event, present these results as for dichotomous data in Subsection 2.5.1, and include relative risks and risk differences with their 95% CIs across the groups for each study, separately. Interpret the results, where appropriate.
Analyse the relative adverse event rates (events per period at risk), if the average period at risk per participant varies substantially between treatment groups (eg using a straight Poisson regression or a negative binomial approach). Present the assumptions associated with statistical analyses and how they were tested.
See Subsection 2.7 for further discussion of adverse reactions reported from other sources.
2.5.3 Cross-references to source documents
For each trial, specify the source document in the reports or papers accompanying the main body of the submission. For each of the responses provided for this subsection, cross-reference the page, table or figure numbers of the relevant trial report(s) (in a separate technical document or attachment, if necessary).
For statistical approaches that are not presented in a clinical study report and cannot be replicated using the data provided in this subsection, present the statistical code (including adequate explanation of covariates) and the statistical outputs in a separate technical document.
